Dynamic properties of NiAl alloy#
In this tutorial, we study the dynamics of an alloy, both in an ordered and disordered phase. The focus will be on multi-component aspects. Note that this tutorial uses a small simulation cell (i.e., few \(\boldsymbol{q}\)-points) and a short molecular dynamics (MD) trajectory in order to limit the runtime. In practice, one needs to tune these parameters as well as the dynasor parameters for optimal results.
For the purpose of this tutorial, we assume that the MD trajectories have been created beforehand using GPUMD. They can be retrieved from Zenodo via
wget https://zenodo.org/records/16887940/files/trajectory-ordered.nc
wget https://zenodo.org/records/16887940/files/trajectory-disordered.nc
We also need the initial structures in order to assign atomic indices to species:
wget https://zenodo.org/records/16887940/files/model-ordered.xyz
wget https://zenodo.org/records/16887940/files/model-disordered.xyz
The same Zenodo record also contains input and other pertinent output files.
Additional information#
If you want to generate your own trajectories you can use this script to generate input structures. You can then use the GPUMD package and this run.in input file. The latter relies on the UNEP model from Nature Communications 15, 10208 (2024) to run MD simulations. The UNEP parameter file can be retrieved from Zenodo via
wget https://zenodo.org/records/10081677/files/UNEP-v1-model-without-ZBL.txt
Please note that the run file employs the optional NetCDF extension of GPUMD. It is possible to use the standard extended-xyz output format (via the dump_exyz keyword), but the execution time will be longer and the trajectory files will be multiple times larger. When using the NetCDF extension the runs should take about 17 to 19 minutes on an A100 or RTX3090 GPU.
[1]:
import numpy as np
from ase.build import bulk
from ase.io import read
from matplotlib import pyplot as plt
from seekpath import get_path
from dynasor import compute_dynamic_structure_factors, Trajectory
from dynasor.qpoints import get_supercell_qpoints_along_path
from dynasor.units import radians_per_fs_to_meV
Set up Trajectory with atom type information#
dynasor supports multiple different trajectory formats via internal readers as well as via MDAnalysis and ase. Here, we use the MDAnalysis reader to parse trajectories in NetCDF format. To begin with, we read the full trajectory for the ordered NiAl (B2 phase, so 50% Ni and 50% Al). The length and time units of the MD trajectory are specified with the length_unit and time_unit keyword.
[2]:
traj = Trajectory('trajectory-ordered.nc',
trajectory_format='nc',
length_unit='Angstrom',
time_unit='fs',
frame_step=1,
frame_stop=None)
INFO 2025-08-25 11:01:47: Trajectory file: trajectory-ordered.nc
INFO 2025-08-25 11:01:47: Total number of particles: 27648
INFO 2025-08-25 11:01:47: Number of atom types: 1
INFO 2025-08-25 11:01:47: Number of atoms of type X: 27648
INFO 2025-08-25 11:01:47: Simulation cell (in Angstrom):
[[68.64 0. 0. ]
[ 0. 68.64 0. ]
[ 0. 0. 68.64]]
A short summary of the content of the traj object can be obtained via the display command (in notebooks) or print (in notebooks or scripts).
[3]:
display(traj)
Trajectory
| Field | Value | File name | trajectory-ordered.nc |
|---|---|
| Number of atoms | 27648 |
| Cell metric | [[68.64 0. 0. ] [ 0. 68.64 0. ] [ 0. 0. 68.64]] |
| Frame step | 1 |
| Atom types | ['X'] |
When we create the Trajectory object in the simplest way, as above, we see that all atoms in the trajectory are taken to be of a single, unknown, type 'X'. We know that this is not the case for our trajectory, since it contains two atom types, 'Ni' and 'Al'.
In order to pass on this information to the Trajectory object, we need to specify the atomic_indices keyword. There are several options the simplest of which is to set atomic_indices='read_from_trajectory'. This requires the species information to be available in the trajectory. While this is the case for, e.g., trajectories in extended-xyz formats, this option is, however, unfortunately not available in the case with these NetCDF trajectories. Here, we therefore prepare a dictionary
the keys of which indicate the species and the values are lists of the corresponding atomic indices.
We note that as a further option you could provide a Gromacs ndx-style index file.
[4]:
structure = read('model-ordered.xyz')
atomic_indices = {s: [a.index for a in structure if a.symbol == s]
for s in set(structure.symbols)}
traj = Trajectory('trajectory-ordered.nc',
trajectory_format='nc',
atomic_indices=atomic_indices,
length_unit='Angstrom',
time_unit='fs',
frame_step=1,
frame_stop=None)
display(traj)
INFO 2025-08-25 11:01:47: Trajectory file: trajectory-ordered.nc
INFO 2025-08-25 11:01:47: Total number of particles: 27648
INFO 2025-08-25 11:01:47: Number of atom types: 2
INFO 2025-08-25 11:01:47: Number of atoms of type Ni: 13824
INFO 2025-08-25 11:01:47: Number of atoms of type Al: 13824
INFO 2025-08-25 11:01:47: Simulation cell (in Angstrom):
[[68.64 0. 0. ]
[ 0. 68.64 0. ]
[ 0. 0. 68.64]]
Trajectory
| Field | Value | File name | trajectory-ordered.nc |
|---|---|
| Number of atoms | 27648 |
| Cell metric | [[68.64 0. 0. ] [ 0. 68.64 0. ] [ 0. 0. 68.64]] |
| Frame step | 1 |
| Atom types | ['Al', 'Ni'] |
Now our Trajectory object contains information about the different atomic types. (N.B.: Since we are not interested in the mass in this case, we can safely ignore the UserWarning issued by MDAnalysis.)
Note that we are not limited to atomic species, but we can also group the atoms in other ways. For example, imagine a system containing bulk and surface atoms for which we would like to study the dynamics separately. In that case we could easily separate these groups by creating a dictionary with the structure dict(bulk=[...], surface=[...]) in the same fashion as above.
Now that we have explored different ways to set up a trajectory containing information about the atom types, let us use the Trajectory object to perform a dynasor calculation and study which atom types in the system give rise to which features in \(S(\boldsymbol{q},\omega)\).
Set up path through Brillouin zone#
We need a path through the Brillouin zone, for which we follow the approach introduced in the “Dynamic properties of FCC-Al” tutorial. In this case, we use the same path through the Brillouin zone for NiAl as for pure Al.
[5]:
repeats = 24 # We simulated a 24x24x24 supercell
prim = bulk('Al', a=traj.cell[0][0] / repeats)
path_info = get_path((prim.cell, prim.get_scaled_positions(), prim.numbers))
point_coordinates = path_info['point_coords']
path = path_info['path']
display(point_coordinates)
display(path)
{'GAMMA': [0.0, 0.0, 0.0],
'X': [0.5, 0.0, 0.5],
'L': [0.5, 0.5, 0.5],
'W': [0.5, 0.25, 0.75],
'W_2': [0.75, 0.25, 0.5],
'K': [0.375, 0.375, 0.75],
'U': [0.625, 0.25, 0.625]}
[('GAMMA', 'X'),
('X', 'U'),
('K', 'GAMMA'),
('GAMMA', 'L'),
('L', 'W'),
('W', 'X')]
[6]:
q_segments = get_supercell_qpoints_along_path(
path, point_coordinates, prim.cell, traj.cell)
q_points = np.vstack(q_segments)
Compute correlation functions#
Now we have prepared everything we need to be able to compute the dynamic structure factor.
Note: The Trajectory object is an “exhaustible iterable” which means that once we have iterated over all frames, it is exhausted. Therefore, one needs to “reload” the trajectory before calling compute_dynamic_structure_factors again to ensure that the entire trajectory is available for analysis.
The code cell below takes about 10 to 15 minutes to run on a common workstation. If you would like to reduce this, set frame_stop above to only read a fraction of the trajectory.
[7]:
%%time
delta_t = 5.0 # MD time step used (fs)
dump_every = 5 # How often the frames were dumped during MD
sample_ordered = compute_dynamic_structure_factors(traj, q_points,
dt=delta_t*dump_every,
window_size=2000,
window_step=100,
calculate_currents=True)
# convert from frequency (rad/fs) to energy (meV) scale
sample_ordered.omega *= radians_per_fs_to_meV
display(sample_ordered)
INFO 2025-08-25 11:01:58: Spacing between samples (frame_step): 1
INFO 2025-08-25 11:01:58: Time between consecutive frames in input trajectory (dt): 25.0 fs
INFO 2025-08-25 11:01:58: Time between consecutive frames used (dt * frame_step): 25.0 fs
INFO 2025-08-25 11:01:58: Time window size (dt * frame_step * window_size): 50000.0 fs
INFO 2025-08-25 11:01:58: Angular frequency resolution: dw = 0.000063 rad/fs = 0.041 meV
INFO 2025-08-25 11:01:58: Maximum angular frequency (dw * window_size): 0.125664 rad/fs = 82.713 meV
INFO 2025-08-25 11:01:58: Nyquist angular frequency (2pi / frame_step / dt / 2): 0.125664 rad/fs = 82.713 meV
INFO 2025-08-25 11:01:58: Calculating current (velocity) correlations
INFO 2025-08-25 11:01:58: Number of q-points: 90
INFO 2025-08-25 11:02:17: Processing window 0 to 2000
INFO 2025-08-25 11:02:28: Processing window 1000 to 3000
INFO 2025-08-25 11:02:39: Processing window 2000 to 4000
INFO 2025-08-25 11:02:49: Processing window 3000 to 5000
INFO 2025-08-25 11:03:00: Processing window 4000 to 6000
INFO 2025-08-25 11:03:11: Processing window 5000 to 7000
INFO 2025-08-25 11:03:22: Processing window 6000 to 8000
INFO 2025-08-25 11:03:32: Processing window 7000 to 9000
INFO 2025-08-25 11:03:43: Processing window 8000 to 10000
INFO 2025-08-25 11:03:54: Processing window 9000 to 11000
INFO 2025-08-25 11:04:05: Processing window 10000 to 12000
INFO 2025-08-25 11:04:16: Processing window 11000 to 13000
INFO 2025-08-25 11:04:26: Processing window 12000 to 14000
INFO 2025-08-25 11:04:37: Processing window 13000 to 15000
INFO 2025-08-25 11:04:48: Processing window 14000 to 16000
INFO 2025-08-25 11:04:59: Processing window 15000 to 17000
INFO 2025-08-25 11:05:10: Processing window 16000 to 18000
INFO 2025-08-25 11:05:20: Processing window 17000 to 19000
INFO 2025-08-25 11:05:31: Processing window 18000 to 19999
INFO 2025-08-25 11:05:33: Processing window 19000 to 19999
DynamicSample
Simulation
| Name | Content |
|---|---|
| Angular frequency resolution | 6.283185307179586e-05 |
| Atom types | ['Al', 'Ni'] |
| Cell | [[68.64 0. 0. ] [ 0. 68.64 0. ] [ 0. 0. 68.64]] |
| Maximum time lag | 50000.0 |
| Number of frames | 20000 |
| Particle counts | {'Ni': 13824, 'Al': 13824} |
| Time between frames | 25.0 |
Dimensions
| Field | Size |
|---|---|
| omega | (2001,) |
| q_points | (90, 3) |
| time | (2001,) |
History
| Function | Field | Content |
|---|---|---|
| compute_dynamic_structure_factors | dt | 25.0 |
| window_size | 2000 | |
| window_step | 100 | |
| calculate_currents | True | |
| calculate_incoherent | False | |
| date_time | 2025-08-25T11:05:37 | |
| username | erhart | |
| hostname | pieweack | |
| dynasor_version | 2.2 |
CPU times: user 25min 33s, sys: 2.03 s, total: 25min 35s
Wall time: 3min 38s
Visualize the dynamic structure factor#
We can now visualize \(S(\boldsymbol{q},\omega)\) using the same approach introduced in the “Dynamic properties of FCC-Al” tutorial, i.e., in the form of a heatmap in the momentum-frequency (or energy) plane. To do so, we first need the running pairwise distance between \(\boldsymbol{q}\)-points along the path.
[8]:
q_distances = []
q_labels = dict()
# starting point
qr = 0.0
# collect labels and q-distances along the entire q-point path
for it, q_segment in enumerate(q_segments):
q_distances.append(qr)
q_labels[qr] = path[it][0]
for qi, qj in zip(q_segment[1:], q_segment[:-1]):
qr += np.linalg.norm(qi - qj)
q_distances.append(qr)
q_labels[qr] = path[-1][1]
q_distances = np.array(q_distances)
First, we visualize the full dynamic structure factor, which, in addition to acoustic branches similar to those we saw in the “Dynamic properties of FCC-Al” tutorial, also reveals optical branches, as expected in a multi-component system. As the dynamic structure factor only contains information about the longitudinal modes, we can in addition visualize the longitudinal and transverse current correlation functions, to study the full dispersion. Here, we plot the difference between the longitudinal and transverse functions, which is a convenient trick to separate the longitudinal (red) and transverse (blue) features visually.
[9]:
fig, axes = plt.subplots(figsize=(5.2, 4.8), nrows=2, dpi=140,
sharex=True, sharey=True)
ax = axes[0]
ax.pcolormesh(q_distances, sample_ordered.omega,
sample_ordered.Sqw_coh.T, cmap='Reds', vmin=0, vmax=8)
ax.text(0.05, 0.85, r'$S(\mathbf{q}, \omega)$', transform=ax.transAxes,
bbox={'color': 'white', 'alpha': 0.8, 'pad': 3})
ax = axes[1]
m = 0.05 * np.max(sample_ordered.Clqw.T - sample_ordered.Ctqw.T)
ax.pcolormesh(q_distances, sample_ordered.omega,
sample_ordered.Clqw.T - sample_ordered.Ctqw.T, cmap='seismic', vmin=-m, vmax=m)
ax.text(0.05, 0.85, r'$C_{L-T}(\mathbf{q}, \omega)$', transform=ax.transAxes,
bbox={'color': 'white', 'alpha': 0.8, 'pad': 3})
for ax in axes:
xticks = []
xticklabels = []
for q_dist, q_label in q_labels.items():
ax.axvline(q_dist, c='0.5', alpha=0.5, ls='--')
xticks.append(q_dist)
xticklabels.append(q_label.replace('GAMMA', r'$\Gamma$'))
ax.set_xticks(xticks)
ax.set_xticklabels(xticklabels)
ax.set_xlim([0, q_distances.max()])
ax.set_ylim([0, 55])
ax.set_ylabel('Energy (meV)', y=1)
fig.tight_layout()
fig.subplots_adjust(hspace=0)
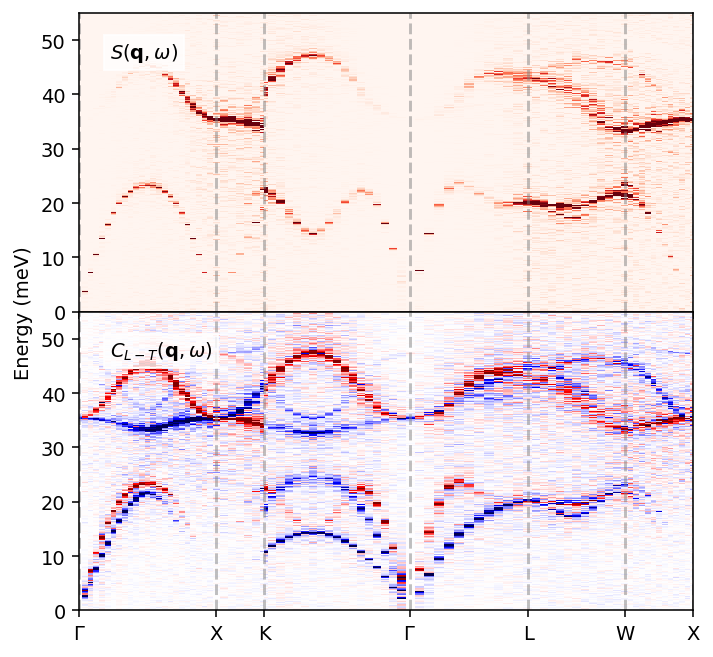
Note again that these correlations are not converged and that one in practice would need to tailor the simulation cell size, the trajectory length, as well as dynasor parameters (window_size, window_step, etc.) to reach convergence.
Since we have added information about atom types to the trajectory, our Sample object also contains the partial structure factors and current correlations, which we can visualize. By doing so, we can learn more about which features in the dispersion relation originate from which atom types in the system.
[10]:
fig, axes = plt.subplots(figsize=(5.2, 6), nrows=3, dpi=140,
sharex=True, sharey=True)
ax = axes[0]
m = 0.05 * np.max(sample_ordered.Clqw_Al_Al.T + sample_ordered.Ctqw_Al_Al.T)
ax.pcolormesh(q_distances, sample_ordered.omega,
sample_ordered.Clqw_Al_Al.T + sample_ordered.Ctqw_Al_Al.T, cmap='seismic', vmin=-m, vmax=m)
ax.text(0.05, 0.85, r'$C_{AlAl}(\mathbf{q}, \omega)$', transform=ax.transAxes,
bbox={'color': 'white', 'alpha': 0.8, 'pad': 3})
ax = axes[1]
m = 0.05 * np.max(sample_ordered.Clqw_Al_Ni.T + sample_ordered.Ctqw_Al_Ni.T)
ax.pcolormesh(q_distances, sample_ordered.omega,
sample_ordered.Clqw_Al_Ni.T + sample_ordered.Ctqw_Al_Ni.T, cmap='seismic', vmin=-m, vmax=m)
ax.text(0.05, 0.85, r'$C_{NiAl}(\mathbf{q}, \omega)$', transform=ax.transAxes,
bbox={'color': 'white', 'alpha': 0.8, 'pad': 3})
ax = axes[2]
m = 0.05 * np.max(sample_ordered.Clqw_Ni_Ni.T + sample_ordered.Ctqw_Ni_Ni.T)
ax.pcolormesh(q_distances, sample_ordered.omega,
sample_ordered.Clqw_Ni_Ni.T + sample_ordered.Ctqw_Ni_Ni.T, cmap='seismic', vmin=-m, vmax=m)
ax.text(0.05, 0.85, r'$C_{NiNi}(\mathbf{q}, \omega)$', transform=ax.transAxes,
bbox={'color': 'white', 'alpha': 0.8, 'pad': 3})
for ax in axes:
xticks = []
xticklabels = []
for q_dist, q_label in q_labels.items():
ax.axvline(q_dist, c='0.5', alpha=0.5, ls='--')
xticks.append(q_dist)
xticklabels.append(q_label.replace('GAMMA', r'$\Gamma$'))
ax.set_xticks(xticks)
ax.set_xticklabels(xticklabels)
ax.set_xlim([0, q_distances.max()])
ax.set_ylim([0, 55])
axes[1].set_ylabel('Energy (meV)')
fig.tight_layout()
fig.subplots_adjust(hspace=0)
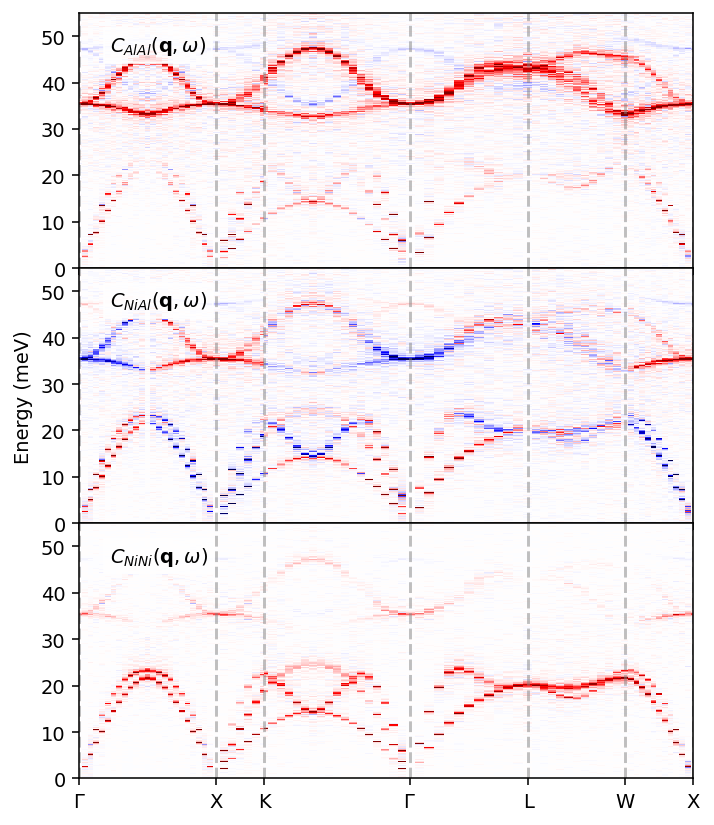
We can here clearly see which atom types contribute to which parts of the dispersion relations, as well as note that some parts have negative sign (orange), in particular for the NiAl partial current correlation.
Dynamics in disordered NiAl#
Now we want to see what changes in the dynamics of disordered NiAl compared to ordered NiAl. To this end, we can use the same procedure as above and compute the correlations, using the same path through the Brillouin zone to be able to compare the two.
[11]:
structure = read('model-disordered.xyz')
atomic_indices = {s: [a.index for a in structure if a.symbol == s] for s in set(structure.symbols)}
traj_disordered = Trajectory('trajectory-disordered.nc',
trajectory_format='nc',
atomic_indices=atomic_indices,
length_unit='Angstrom',
time_unit='fs',
frame_step=1,
frame_stop=None)
INFO 2025-08-25 11:05:38: Trajectory file: trajectory-disordered.nc
INFO 2025-08-25 11:05:38: Total number of particles: 27648
INFO 2025-08-25 11:05:38: Number of atom types: 2
INFO 2025-08-25 11:05:38: Number of atoms of type Ni: 13824
INFO 2025-08-25 11:05:38: Number of atoms of type Al: 13824
INFO 2025-08-25 11:05:38: Simulation cell (in Angstrom):
[[68.64 0. 0. ]
[ 0. 68.64 0. ]
[ 0. 0. 68.64]]
Again, the code cell below takes about 10 to 15 minutes to run on a common workstation. If you would like to reduce this, set frame_stop above to only read in a fraction of the trajectory.
[12]:
delta_t = 5.0 # MD time step used (fs)
dump_every = 5 # How often the frames were dumped during MD
sample_disordered = compute_dynamic_structure_factors(traj_disordered, q_points,
dt=delta_t*dump_every,
window_size=2000,
window_step=100,
calculate_currents=True)
sample_disordered.omega *= radians_per_fs_to_meV # convert from frequency (rad/fs) to energy (meV) scale
INFO 2025-08-25 11:05:38: Spacing between samples (frame_step): 1
INFO 2025-08-25 11:05:38: Time between consecutive frames in input trajectory (dt): 25.0 fs
INFO 2025-08-25 11:05:38: Time between consecutive frames used (dt * frame_step): 25.0 fs
INFO 2025-08-25 11:05:38: Time window size (dt * frame_step * window_size): 50000.0 fs
INFO 2025-08-25 11:05:38: Angular frequency resolution: dw = 0.000063 rad/fs = 0.041 meV
INFO 2025-08-25 11:05:38: Maximum angular frequency (dw * window_size): 0.125664 rad/fs = 82.713 meV
INFO 2025-08-25 11:05:38: Nyquist angular frequency (2pi / frame_step / dt / 2): 0.125664 rad/fs = 82.713 meV
INFO 2025-08-25 11:05:38: Calculating current (velocity) correlations
INFO 2025-08-25 11:05:38: Number of q-points: 90
INFO 2025-08-25 11:06:01: Processing window 0 to 2000
INFO 2025-08-25 11:06:14: Processing window 1000 to 3000
INFO 2025-08-25 11:06:26: Processing window 2000 to 4000
INFO 2025-08-25 11:06:39: Processing window 3000 to 5000
INFO 2025-08-25 11:06:52: Processing window 4000 to 6000
INFO 2025-08-25 11:07:05: Processing window 5000 to 7000
INFO 2025-08-25 11:07:18: Processing window 6000 to 8000
INFO 2025-08-25 11:07:31: Processing window 7000 to 9000
INFO 2025-08-25 11:07:43: Processing window 8000 to 10000
INFO 2025-08-25 11:07:56: Processing window 9000 to 11000
INFO 2025-08-25 11:08:09: Processing window 10000 to 12000
INFO 2025-08-25 11:08:23: Processing window 11000 to 13000
INFO 2025-08-25 11:08:37: Processing window 12000 to 14000
INFO 2025-08-25 11:08:52: Processing window 13000 to 15000
INFO 2025-08-25 11:09:06: Processing window 14000 to 16000
INFO 2025-08-25 11:09:20: Processing window 15000 to 17000
INFO 2025-08-25 11:09:35: Processing window 16000 to 18000
INFO 2025-08-25 11:09:49: Processing window 17000 to 19000
INFO 2025-08-25 11:10:03: Processing window 18000 to 19999
INFO 2025-08-25 11:10:05: Processing window 19000 to 19999
[13]:
fig, axes = plt.subplots(figsize=(5.2, 4.8), nrows=2, dpi=140,
sharex=True, sharey=True)
ax = axes[0]
ax.pcolormesh(q_distances, sample_disordered.omega,
sample_disordered.Sqw_coh.T, cmap='Reds', vmin=0, vmax=5)
ax.text(0.05, 0.85, r'$S(\mathbf{q}, \omega)$', transform=ax.transAxes,
bbox={'color': 'white', 'alpha': 0.8, 'pad': 3})
ax = axes[1]
m = 0.3 * np.max(sample_disordered.Clqw.T - sample_disordered.Ctqw.T)
ax.pcolormesh(q_distances, sample_disordered.omega,
sample_disordered.Clqw.T - sample_disordered.Ctqw.T, cmap='seismic',
vmin=-m, vmax=m)
ax.text(0.05, 0.85, r'$C_{L-T}(\mathbf{q}, \omega)$', transform=ax.transAxes,
bbox={'color': 'white', 'alpha': 0.8, 'pad': 3})
for ax in axes:
xticks = []
xticklabels = []
for q_dist, q_label in q_labels.items():
ax.axvline(q_dist, c='0.5', alpha=0.5, ls='--')
xticks.append(q_dist)
xticklabels.append(q_label.replace('GAMMA', r'$\Gamma$'))
ax.set_xticks(xticks)
ax.set_xticklabels(xticklabels)
ax.set_xlim([0, q_distances.max()])
ax.set_ylim([0, 55])
ax.set_ylabel('Energy (meV)', y=1)
fig.tight_layout()
fig.subplots_adjust(hspace=0)
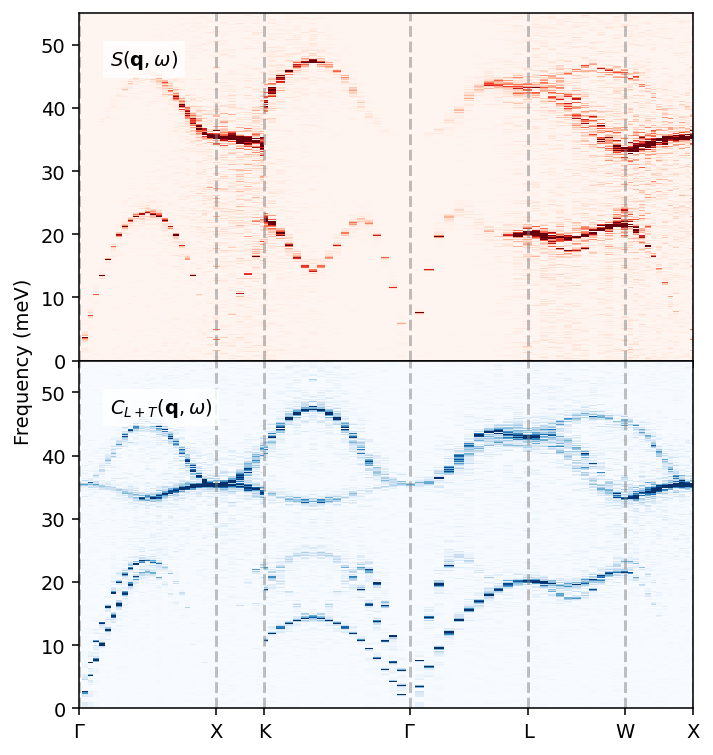
With the dynamic structure factors and current correlation at hand for both the ordered (B2) and the disordered system, we can readily compare the two, and note the smearing of the bands that arises in the disordered case. One might consider averaging the disordered case over \(|\boldsymbol{q}|\) instead, following the approach commonly used for liquids.